Command Palette
Search for a command to run...
Achieving Highly Selective Substrate Design, MIT and Harvard Collaborate to Discover Novel Protease Cleavage Patterns Using Generative AI.

In the intricate biochemical reaction network of living organisms, proteases can specifically cleave peptide bonds, thereby precisely regulating a series of key life processes, from blood clotting and tissue repair to immune responses and even cancer progression. Dysfunction of these proteases often directly leads to the occurrence and development of various serious diseases. Therefore, elucidating the mechanisms of action of proteases and precisely regulating their activity is not only a core issue in basic life sciences but also a crucial breakthrough for developing novel diagnostic and therapeutic methods.
The key to achieving this goal,The key lies in finding peptide substrates that are highly "matched".They can be used as molecular probes to track enzyme activity, designed as inhibitors to block abnormal activity, or even act as "conditional activation switches" in drug delivery systems to achieve targeted therapy.
However, designing peptide substrates that are both rapidly cleaved by target proteases and highly selective (recognized only by that enzyme, avoiding cross-reactions with other proteases) has always been a major challenge for the scientific community. This problem stems from the complex biochemical interactions between proteases and substrates: to adapt to diverse physiological functions, proteases have evolved broad cleavage specificities, and their active sites must bind precisely to peptide substrates (typically about ten amino acids long). Even considering only synthetic peptides of 10 amino acids, using the 20 common natural amino acids, the theoretical sequence combinations can reach approximately 20¹⁰ (nearly 10¹³), creating a nearly infinite space for exploration. Further complicating matters is…Proteases with similar functions often originate from a common ancestor and have similar active site structures, making them highly susceptible to "cross-recognition."This makes it particularly difficult to screen for highly specific substrates from a vast number of possibilities.
To overcome this bottleneck, researchers have made numerous attempts. Traditional methods often rely on known cleavage sites or enzymatic information of natural proteins, resulting in low efficiency and difficulty in obtaining ideal artificial substrates. Rational design based on chemical biology knowledge is usually cumbersome, has limited throughput, and is mostly targeted at single proteases, making it difficult to scale up. In recent years, although high-throughput screening technologies have improved efficiency to some extent, they still suffer from limitations such as complex operation and high cost.Most existing computational prediction methods can only determine "whether to cut" and cannot accurately sort cutting efficiency, thus failing to meet the needs of in-depth mechanism research and engineering applications.
In this context,MIT and Harvard University jointly proposed CleaveNet, an end-to-end design flow based on artificial intelligence.By working in synergy between predictive and generative models, this approach aims to revolutionize the existing paradigm of protease substrate design and provide entirely new solutions for related basic research and biomedical development.

Paper address:
https://www.nature.com/articles/s41467-025-67226-1
Follow our official WeChat account and reply "CleaveNet" in the background to get the full PDF.
More AI frontier papers:
Cross-scenario validation that empowers the generalization ability of the CleaveNet model, supported by datasets from multiple experimental scenarios.
In developing and validating the CleaveNet model, this study integrated two datasets that differed significantly in sequence composition and experimental methods to ensure the model's reliability and generalization ability.
The core dataset used by the researchers came from a published study that systematically characterized the cleavage activity of a substrate library containing approximately 18,500 synthetic decapeptides against 18 matrix metalloproteinases (MMPs) using mRNA display technology.Each substrate-protease combination corresponds to a standardized cleavage efficiency score (Zₛₘ) to quantify the relative cleavage intensity.
To further ensure the rigor of the assessment and avoid overestimation due to sequence similarity,The researchers performed homology filtering on the initial test set:The researchers calculated the minimum Levenstein distance between each test sequence and all sequences in the training set, and removed 816 sequences with a distance less than 3 that were highly similar to the training set. Ultimately, they obtained a "mRNA display test set" containing 2,901 non-overlapping sequences. This subset was not used at any stage of model training and was dedicated solely to internal performance validation.
To independently test the model's adaptability when faced with drastically different biochemical backgrounds,The study also introduced a completely independent out-of-distribution dataset called the "fluorescence test set".This dataset contains 71 synthetic peptides of varying lengths (7-14 amino acids), whose cleavage activity has been validated for seven recombinant MMP proteins using classic in vitro experiments based on fluorescence resonance energy transfer (FRET). This dataset differs fundamentally from the core dataset generated using mRNA display technology in terms of peptide length distribution, amino acid composition, and, most importantly, the experimental detection principles. This intentional design provides a crucial benchmark for evaluating the CleaveNet model's ability to transcend specific experimental conditions and capture universal biochemical patterns.
CleaveNet predicts and generates collaborative closed loops.
As shown in the figure below, the core of CleaveNet consists of two complementary and collaborative computational modules: the prediction module (CleaveNet Predictor) and the generation module (CleaveNet Generator).Together, they form a complete "design-evaluation" closed loop.
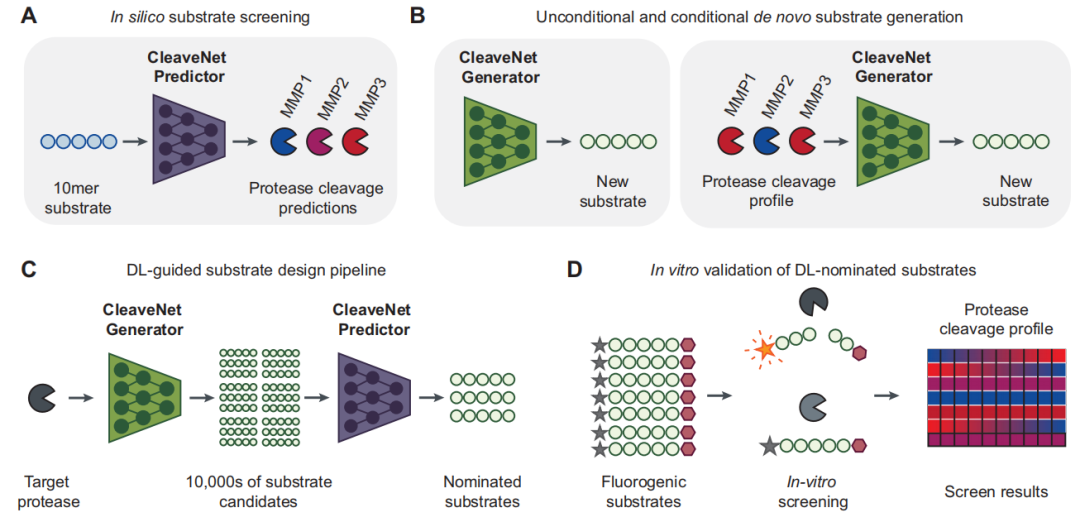
The prediction module aims to solve the problem of rapidly and accurately evaluating the cleavage activity of candidate substrates from a massive sequence space.The researchers constructed it as a multi-output sequence-function regression model. Specifically, the model takes an amino acid sequence as input, and its core task is to simultaneously output the predicted cut score (Ŵₛₘ) of the sequence for all 18 MMPs, and simultaneously estimate the uncertainty (σₛₘ) of each prediction.
To achieve higher predictive robustness, this study employed a model ensemble strategy:Five identical prediction models were independently trained on the mRNA display training set, with the final prediction score being the mean of their outputs. The uncertainty of the predictions was quantified by the standard deviation of these five results. Furthermore, by setting an adjustable threshold (Zₜ), the model can easily convert continuous prediction scores into a binary judgment of "cut" or "uncut," thus serving different screening scenarios.
In constructing the predictive model, this study systematically compared two mainstream architectures in sequence modeling—bidirectional long short-term memory networks and Transformers. The former excels at capturing sequence dependencies, while the latter, with its attention mechanism, can globally model interactions between amino acids and is currently the mainstream choice for protein language representation. Based on its potential demonstrated on larger-scale, more diverse data,The researchers ultimately chose the Transformer architecture as the basis for the CleaveNet Predictor.
The goal of the generation module is to achieve automated and intelligent design of candidate substrates.This study trained a generative model based on an autoregressive Transformer, enabling it to learn the universal MMP cutting preferences inherent in the dataset from mRNA representation.This model can generate a large number of novel and reasonable peptide sequences without any additional input conditions.
In order to scientifically evaluate the value of generative models rather than simply reproduce randomness, researchers have developed a robust baseline method called "site-independent control".This method only counts the independent distribution of each amino acid position in the training data, and then performs random sampling based on this to generate sequences.By comparing the CleaveNet-generated sequences with this baseline sequence across multiple dimensions, we can clearly identify the complex biochemical patterns learned by the model that go beyond simple statistical associations.
The close collaboration between the prediction and generation modules enables researchers to first generate a diverse candidate library, and then perform efficient and accurate virtual screening on it, thus providing a powerful computational engine for subsequent experimental verification.
CleaveNet enables selective and precise control.
After completing the model construction, this study conducted multi-level and systematic experimental verification of CleaveNet's performance, and the results fully demonstrated the outstanding value of this process in terms of prediction accuracy, generation rationality, and practical application effectiveness.
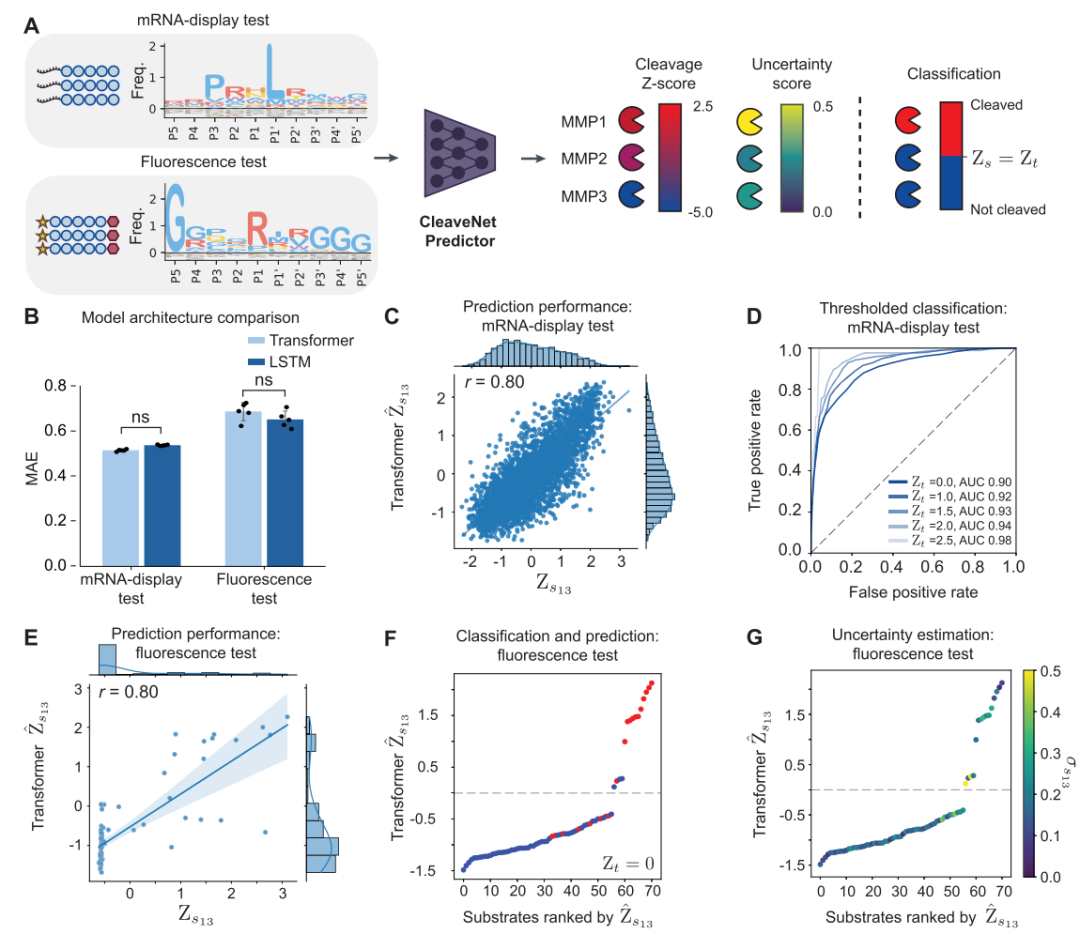
first,CleaveNet Predictor demonstrates excellent predictive capabilities on both internal and external test sets.On a homology filtering test set (mRNA display test set) that was never used in training, the model's predicted score (Ŵₛₘ) for MMP13 showed a strong correlation with the experimentally measured standardized Z score (Zₛₘ) (Pearson correlation coefficient r = 0.80). Its performance was equally robust when the continuous predictions were converted to a "cut/uncut" binary classification: by plotting receiver operating characteristic (ROC) curves and calculating the area under the curve (AUC), researchers found that the model maintained high discriminative power across different decision thresholds, especially at the universally accepted cut threshold (Zₜ=2.5), where the AUC reached 0.98. More rigorous testing came from completely independent fluorescence test sets with vastly different experimental methods.
Despite the fact that the sequence length, amino acid composition, and detection principle of this dataset are different from those of the training data, the model's predicted cut score still maintains a strong positive correlation with the experimental value (r = 0.80 for MMP13), and can accurately distinguish between the experimentally verified "cut" and "uncut" sequences.This strongly confirms that CleaveNet Predictor can not only memorize training data patterns, but also capture the universal biochemical laws governing substrate cleavage by proteases.It has strong generalization ability.
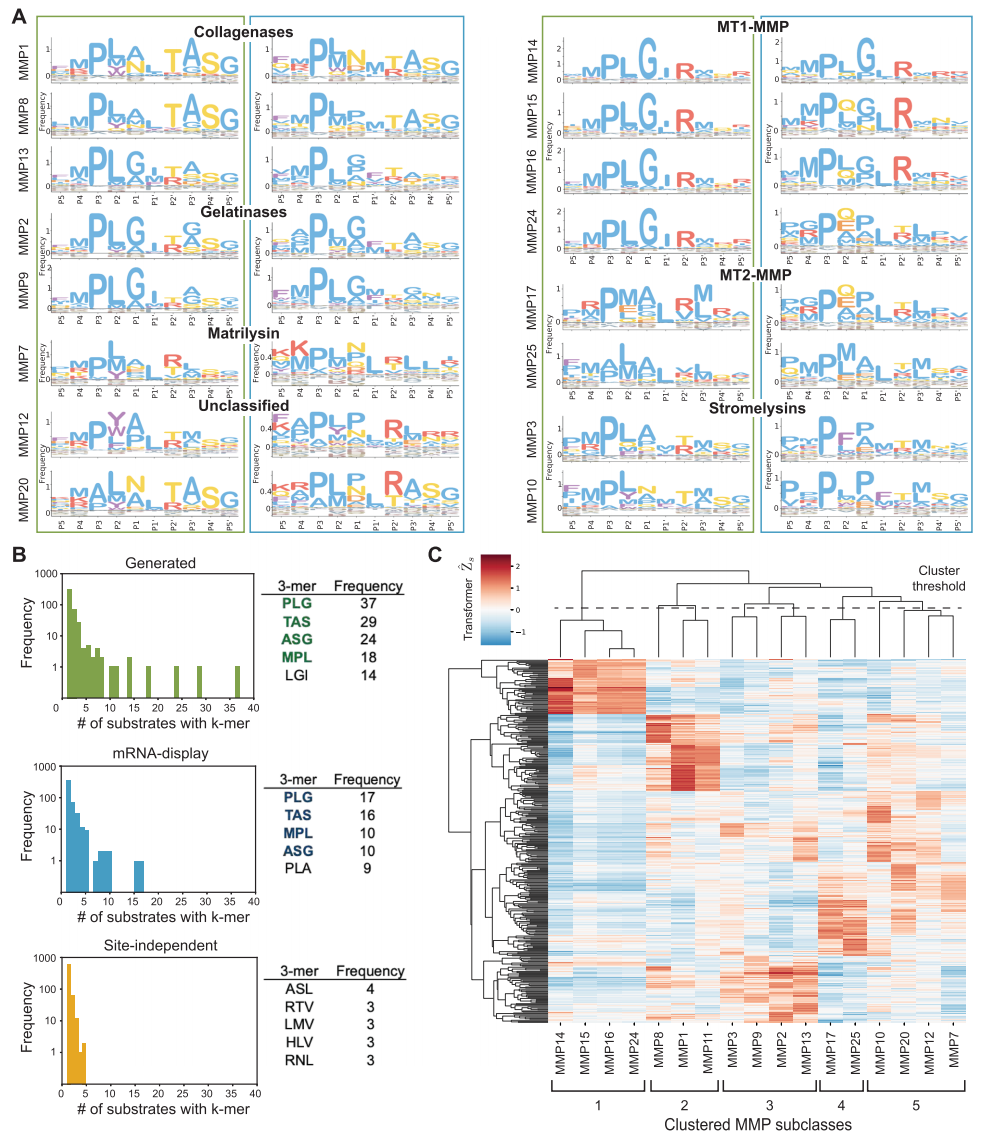
Secondly,Bioinformatics analysis of the sequences generated by CleaveNet Generator by researchers revealed its rationale and novelty.Compared to "site-independent control" sequences based solely on random sampling of single amino acid position frequencies, the sequences generated by the generative model more accurately reproduce the classic cleavage motifs of the MMP family and exhibit an amino acid distribution in key substrate-binding pocket regions that more closely resembles actual experimental data. More importantly,The generated sequences are consistent with the real dataset in terms of overall biophysical properties (such as hydrophobicity and charge).However, high-quality generation does not mean a simple replication of the training data. Sequence diversity analysis showed that the proportion of unique long synthetic peptides shared by the generated sequences and the training set was extremely low, indicating that the model avoided overfitting and was able to explore new sequence spaces not covered by the training data.
Further functional clustering analysis revealed that the predicted cleavage activity spectra of high-scoring substrates generated by different MMPs could be naturally clustered based on the phylogenetic relationships of the MMP catalytic domains.This demonstrates that the generative model not only learns the apparent sequence patterns, but also intrinsically captures information about the functional differentiation in the evolution of proteases.This demonstrates the biological rationality of its generated results.
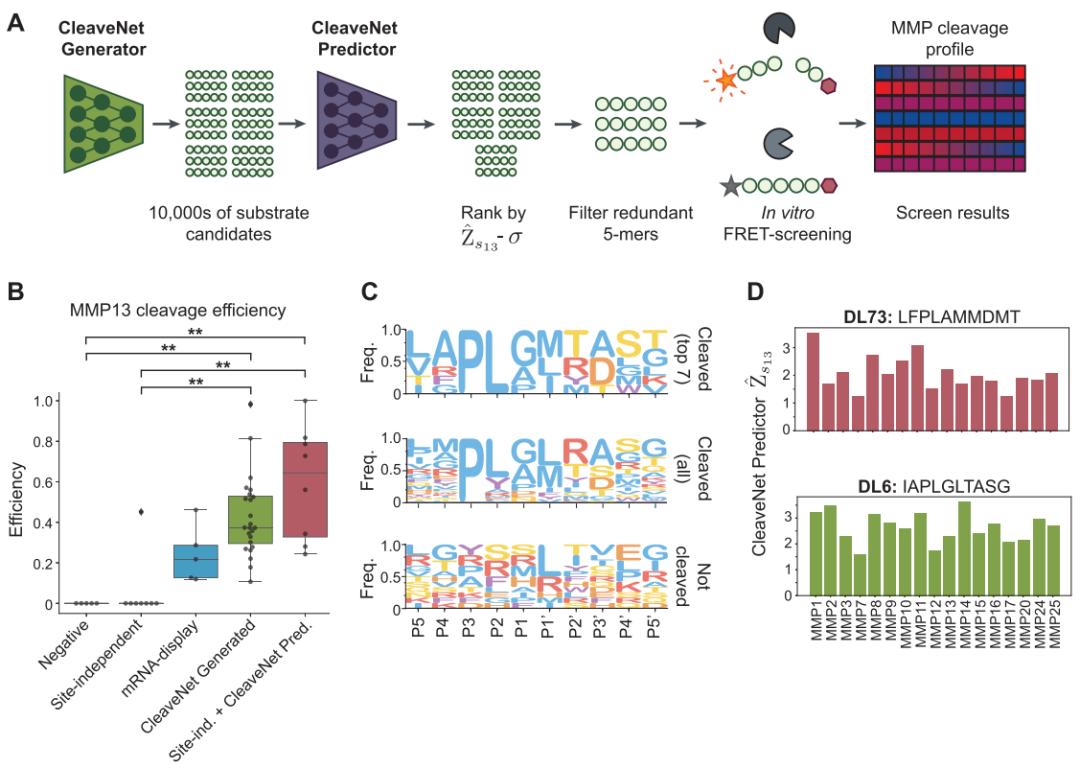
Ultimately, the validity of all computational designs was validated through in vitro biochemical experiments. Researchers synthesized multiple sets of candidate substrates designed by CleaveNet targeting MMP13, including sequences directly generated by the generative model and sequences screened by the predictive model. Fluorescence resonance energy transfer (FRET) cleavage experiments yielded convincing results.All 24 substrates designed using the CleaveNet pipeline were successfully cleaved by the reconstructed MMP13, achieving a hit rate of 100% (TP3T).Furthermore, the median cutting efficiency was significantly higher than that of the known high-efficiency positive control substrates in the training set. This verifies the ability of this process to design high-efficiency substrates.
To demonstrate the potential of the process in tackling more challenging tasks, such as designing highly selective substrates, this study further employed a conditional generation strategy, specifying "high MMP13 selectivity" as the objective in the generative model. Subsequent large-scale parallel in vitro screening (95 substrate pairs for 12 different MMPs) showed that the substrates generated through conditional guidance exhibited high selectivity.The cleavage activity is significantly biased towards MMP13, resulting in higher selectivity.
Of particular note is that some of the designed substrates possess both high cutting efficiency and high selectivity, an excellent combination that is extremely rare in the original training data, highlighting CleaveNet's powerful ability to explore new and high-quality sequence spaces.
In summary, from accurate computational prediction to reasonable sequence generation, and then to conclusive wet experimental verification, a series of interconnected results demonstrate that CleaveNet has constructed an efficient, reliable, and powerful protease substrate design platform. This research not only provides an innovative AI solution to the classic challenge of protease activity regulation but also lays a new methodological foundation for future protease function research and related drug development.
AI-driven innovation in protease substrate design
CleaveNet's AI-assisted protease substrate design technology is driving innovation in the life sciences and biomedicine fields worldwide.
David Baker's team at the University of Washington published groundbreaking research in Science.For the first time, AI has been used to design a serine hydrolase with a complex active site from scratch—one of the largest known families of enzymes.The study introduced a novel machine learning network, PLACER, which not only successfully designed an active enzyme that can efficiently catalyze ester hydrolysis, but also unexpectedly discovered five new protein folding patterns, significantly expanding the structural diversity of this enzyme family.
* Paper Title: Computational design of serine hydrolases
* Paper link:
https://www.science.org/doi/10.1126/science.adu2454
Furthermore, a joint team from several European universities has developed a general model based on the Transformer architecture that can accurately predict protease-substrate interactions. This model integrates global multi-source protease cleavage data, achieving effective prediction of substrate sequences across species. Its generalization ability has been validated in research on proteases from various pathogens, including bacteria and viruses, providing important sequence design basis for the development of anti-infective drugs.
It is foreseeable that, with the continued convergence of computational biology, artificial intelligence, and synthetic biology, protease substrate design will evolve from a science that combines art and experience into a highly rationalized and engineered research field. This will not only accelerate the development of novel drugs, diagnostic tools, and green biocatalysts, but also potentially help us ultimately decode the underlying logic of life regulation, ushering in a new era of on-demand programming of life functions.








